Shotgun Metagenome
Shotgun Metagenome
1. Shotgun Metagenome 분석의 목표
- 환경 샘플내의 어떤 미생물이 구성되며 기능적으로 어떤 역할을 하는지 분석.
- 샘플 내의 다양한 유전체 정보를 재구성, 미생물의 분류체계와 커뮤니티 구조의 특성을 확인, 커뮤니티의 생물학적 기능을 확인.
- Taxonomic classification, Population analysis, Functional analysis.
1) Shotgun metagenome analysis workflow
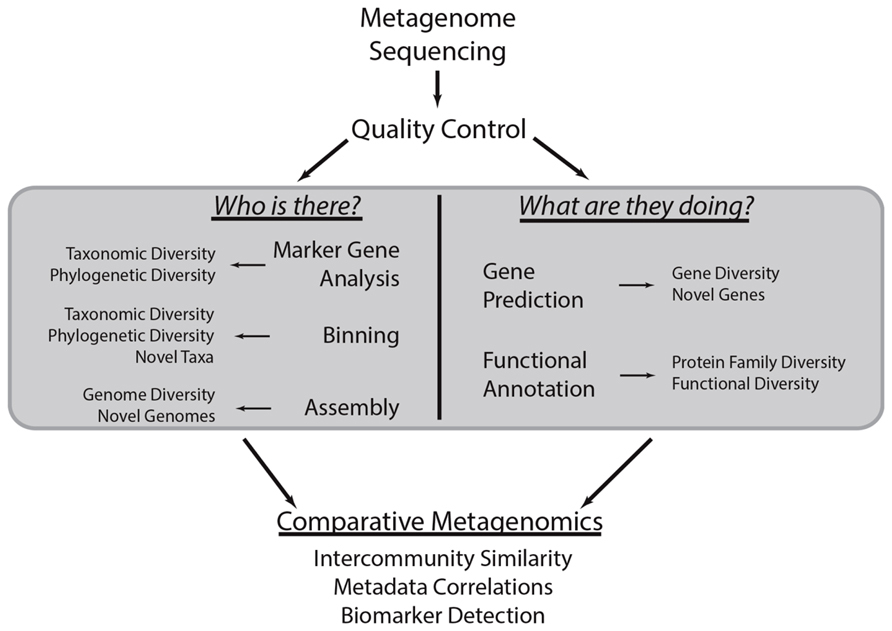

2) NGS 시퀀싱 및 데이터 품질검사
- NGS 플랫폼 중에서 Illumina 기기를 주로 이용함.
2. Shotgun sequencing library 구축
- 무작위로 DNA를 절단한 후 얻은 짧은 서열을 얻어낸 뒤 이를 통해 consensus sequence를 얻어내는 방식.
- FASTAQ format으로 데이터가 나옴.
- Cloning 방법이 아닌 ligation 방법과 amplification 방법을 통해 library 구축.
3. 데이터 품질검사
1) Adapter trimming
- Adapter는 임의적으로 붙인 합성 sequence로 분석 시 분류학적 식별을 복잡하게 하기 때문에, amplicon sequence만 남도록 제거 필요
2) Quality trimming, Quality check
- 품질 기준을 만족하지 않는 모든 서열은, quality trimming이라 불리는 공정을 통해, 추가 분석에서 제거됨.
(서열의 길이, 최소 quality score, 바코드 서열의 불일치 등의 다양한 파라미터 사용) - 낮은 퀄리티의 서열을 제거
- Q20 – Q30 이상을 권장
4. Marker gene analysis
- Marker gene analysis는 metagenome의 taxonomic diversity를 정량화하는 가장 간단하고 효율적인 방법 중 하나이다.
- 분석 과정에서 Metagenomic read를 taxonomically informative gene families (marker genes)의 database와 비교하고 Marker gene homologs를 식별한 뒤, Sequence나 phylogenetic similarity를 사용해 각 metagenomic homolog를 분류학적으로 annotation을 단다.
- 가장 많이 사용되는 marker genes로는 rRNA genes 또는 protein coding genes가 있으며 이들은 single copy이면서 microbial genomes에서 공통적으로 가지고 있다는 특징이 있다.
- 비교적 작은 database와 비교하는 방법으로 비교적 빠른 방법이다.
- Marker genes를 사용해 분류학적으로 metagenome에 annotation을 하는 방법에는 2가지 방법이 있다.
1) Using sequence similarity between the read and marker genes
- 대표적인 tool로 MetaPhler와 MetaPhlAn이 있다.
- MetaPhyler는 marker genes를 taxonomic reference로 사용하는 taxonomic profiler tool로 family (rate of evolution 등)와 read (sequence length 등) property를 고려해 metagenomic sequence의 분류를 결정한다.
- MetaPhlAn은 shotgun metagenomic reads를 input으로 받아 상대적 풍부도와 관련해 species rank에 따라 다른 분류학적 계통군으로 분류된 미생물 목록을 만든다.
- MetaPhyler는 marker genes를 taxonomic reference로 사용하는 taxonomic profiler tool로 family (rate of evolution 등)와 read (sequence length 등) property를 고려해 metagenomic sequence의 분류를 결정한다.
2) Using phylogenetic information
- 시간이 더 오래 거릴 수 있지만 정확도는 더 높다.
- AMPHORA와 PhyloSiftis, PhylOUT가 주로 사용된다.
- AMPHORA는 hidden Markov models (HMMs)를 사용해 bacteria나 archaea의 sequenced genome 에서 공통적으로 나타나는 계통발생학적으로 informative한 single copy protein-coding genes의 metagenomic homologs를 식별한다.
이후 metagenomic homologs를 포함하는 marker gene phylogeny를 assemble한다.
(이 때 metagenomic homologs는 phylogenetic tree에서 상대적인 위치에 따라 annotation이 달려있다.) - PhyloSiftis는 이와 유사하지만 매우 큰 virus gene family database를 포함한 marker database와 edge PCA를 사용해 커뮤니티 간에 다른 marker genes의 phylogenetic tree에서 특정 계통을 식별한다.
- PhylOTU는 중첩되지 않은 metagenomic 16S homologs를 연결하기 위해 phlyogenetic tree를 사용한다.
5. Assembly
- Genome assembly는 original genome sequence를 재구성하기 위해 특정 read를 더 긴 sequence로 병합하는 과정으로 이상적인 결과는 완전한 genome sequence를 완성하는 것이다.
- 1차적으로 read를 바탕으로 assembly 진행 후 만들어진 더 긴 서열을 contig라고 하며, contig들끼리 assemble하여 유전체 수준의 scaffold를 구성한다.
- Unassembled metagenomic reads에 비해 분석을 단순화할 수 있다.
- 커뮤니티에서 발견되는 미배양 미생물의 genomic composition을 알 수 있다.
5 - 1. Assembly 전략
1) Reference-guided assembly
- 참조 유전체(database)에 정렬하는 방법이다.
- 기존 알려진 참조 유전체 서열과 해당 품질에 제한적이다.
- 아래와 같은 케이스에는 사용하기 어렵다.
- Larger genomic mutations (insertions, deletions, rearrangements)
- Distantly related species (유연관계가 매우 먼 종의 분석)
- Most virus
- Larger genomic mutations (insertions, deletions, rearrangements)
2) De novo assenbly
- 시퀀싱 read의 충분한 lengths, depths, coverage가 필요하다.
- 어떤 알고리즘과 파라미터를 결정하느냐에 따라 결과가 많이 달라진다.
- 정답 sequence를 알지 못하는 상태에서 진행한다.
- Low coverage 영역이나 long repeat 영역에서는 쉽지 않다.
- 알고리즘의 종류
- De Brujin graph
- 전통적인 방법으로 그래프 접근 방식을 기반으로 한다. 그래프는 overlapping sequence identity를 통해 연결된 다른 모든 read의 해당 k-mer 뿐만 아니라 read에서 지정된 길이 (k-mer)의 모든 하위 sequence간의 연속적인 sequence overlap을 모델링한다. - Overlap-layout-consensus
- Greedy assembly (only to illustrate)
- De Brujin graph
5 - 2. Assembly시 주의사항
- 두 개의 별개 genome의 sequence가 서로 공유된 sequence similarity로 인해 contig로 잘못 조립될 수 있으며 이를 chimera라고 한다. 보통 chimera는 복잡한 커뮤니티에서 생성될 가능성이 더 높다. Chimera의 생성 위험을 완화하기 위해 종종 read를 버리거나 각 bin을 독립적으로 조립한다.
- Assembly는 community에서 가장 풍부한 분류군으로 제한되는 경향이 있다. 따라서 extensive sequencing 없이는 희귀한 미생물의 genome을 assembly하기 어렵다.
6. Binning

- Binning은 모든 metagenomic sequences를 taxonomical group (OTU, genus, family 등)에 할당한다.
- 일반적으로 각 sequence는
- 일부 reference data와 비교하여 taxonomic group으로 분류되거나
- GC content와 같은 공유하는 특성을 기반으로 taxonomic groups를 나타내는 sequence group으로 clustering된다.
- 일부 reference data와 비교하여 taxonomic group으로 분류되거나
- Binning 과정은 amplicon 데이터 처리에서 OTU clustring과 동일하며 미생물 군집의 taxonomic diversity를 특성화하는 역할을 한다.
- Binning의 이점 3가지
- 사용된 방법에 따라 다른 방법으로는 식별하기 어려운 새로운 genome의 존재를 찾아낼 수 있다.
- Community에서 구별되는 분류군의 수와 유형에 대해 알 수 있다.
- Binning 후 분석(assembly 등)이 전체 데이터 모집단이 아닌 각 binning된 read set에 대해 독립적으로 수행될 수 있도록 데이터의 복잡성을 줄이는 방법을 제공한다.
- 사용된 방법에 따라 다른 방법으로는 식별하기 어려운 새로운 genome의 존재를 찾아낼 수 있다.
- Binning은 분류법에 의존하는 방법과 독립적으로 분석하는 방법으로 나뉜다.
- 최신 metagenome binning의 경향은 taxonomy independent 형태이다
6 - 1) Taxonomy independent binning
- Reference database가 불완전하기 때문에 새로운 binning 알고리즘의 대부분은 reference-free 접근 방식이다.
- Composition based methods와 abundance based methods, hybrid methods로 나뉜다.
1) Composition based methods
- Genome composition은 각 분류군에서 고유하므로 이를 비교하면 순전히 sequence를 binnig할 수 있다는 가정에 기반한 방법이다.
- Sequence 구성은 character based이기 때문에 먼저 적절한 numerical feature vector로 변환해야하며 이 때 가장 많이 사용되는 것이 genomic signatures와 GC contents이다.
(Genomic signatures: 특정 크기의 k-mer의 정규화된 빈도수) - 문제점
1. Abnundance가 낮은 종의 sequence의 경우 작고 불분명한 cluster를 형성하며 abundance가 높은 종에 속하는 보다 큰 bin의 일부로 쉽게 오분류될 수 있다.
2. 긴 sequence (e.g. 800bp)를 사용할 때만 합리적으로 정확한 결과를 제공한다.
2) Abundance based methods
- 미생물 샘플에서 taxon의 abundance를 반영하는 contig coverage를 기반으로 한다.
- Composition based methods에서 나타난 문제점을 해결할 수 있다.
- 낮은 abundance의 경우 하나의 sample로 작업하는 방법인 AbundanceBin과 MBBC로 해결이 가능하다.
- AbundanceBin의 핵심은 포아송 분포를 적용해 각 nucleotide의 적용 범위를 계산할 수 있을 때, sequencing된 read의 분포가 Lander-Waterman model을 따른다는 것이다.
이러한 방법의 workflow는 composition based binning 기술과 다소 유사하며 cluster 형성의 주요 차이점은 유사성(composition) 대신 k-mer abundance로 정의된다는 것이다. - MBBC는 동일한 genome에서 나온 contig의 coverage profiles가 여러 샘플에서 높은 상관관계가 있어야 한다는 가정을 기반으로 한다.
- AbundanceBin의 핵심은 포아송 분포를 적용해 각 nucleotide의 적용 범위를 계산할 수 있을 때, sequencing된 read의 분포가 Lander-Waterman model을 따른다는 것이다.
- 긴 sequence를 사용해야만 하는 문제점은 AbundanceBin을 통해 해결이 가능하다.
- AbundanceBin은 75bp 정도의 짧은 길이에서도 정확하게 작동할 수 있다.
- 낮은 abundance의 경우 하나의 sample로 작업하는 방법인 AbundanceBin과 MBBC로 해결이 가능하다.
3) Hybrid methods
- Composition based binning과 abundance based binning을 합친 방법이다.
- Sequence composition과 coverage 정보를 조합할 경우 metagenomics data에 대한 더 많은 정보를 추출할 수 있으며 이는 더 정확한 binning 결과로 이어진다는 것이 입증되었다.
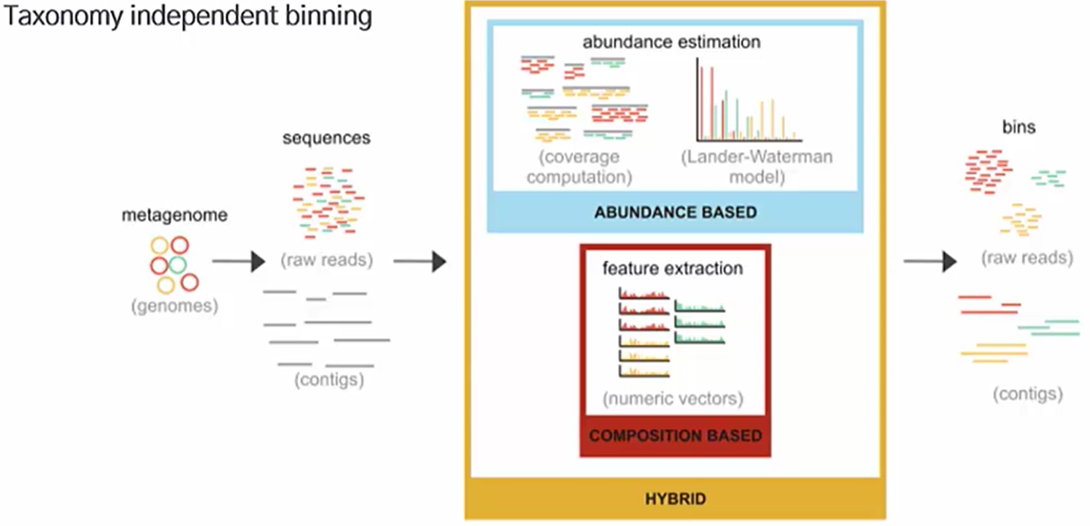

6 - 2) Binnig시 주의사항
- 일반적으로 binning된 read 수와 각 bin에 할당된 annotation의 분류학적 특이성 사이에 절충점이 있다. 또한 binning은 metagenome의 상당 부분에 annotation을 달 수 있는 방법을 제공하지만 read 전반에 걸쳐 annotation의 정확도와 특이성에 큰 차이가 있을 수 있다.
- Horizontal gene transfer를 포함한 convergent evolutional characteristic은 특히 composition based method와 train data로 잘 표현되지 않을 수 있는 분류군 연구에서 binning의 정확도를 감소시킬 수 있다.
- 새로운 유기체의 경우 알고리즘의 예측을 검증하는 것이 어렵다.
7. Inferring Biological function
- Community의 functional diversity는 metagenomic sequence에 function을 annotating하여 정량화할 수 있다.
- 일반적으로 단백질 coding sequence를 포함하는 metagenomic read를 식별하고 functional information을 알고있는 genes, proteins, protein families, 또는 metabolic pathways의 database와 coding sequence를 비교하는 것을 포함한다.
- Biological function은 database sequence와의 similarity를 기반으로 추론된다.
- 일반적으로 단백질 coding sequence를 포함하는 metagenomic read를 식별하고 functional information을 알고있는 genes, proteins, protein families, 또는 metabolic pathways의 database와 coding sequence를 비교하는 것을 포함한다.
- Metagenome은 새로운 유전자를 발견하거나 biological function이 아직 알려지지 않은 유전자와 관련된 ecological condition에 대한 정보를 제공할 수 있다.
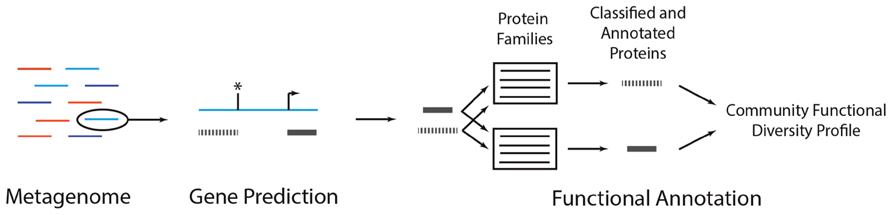
7 - 1) Gene prediction
- Metagenome에서 Gene prediction에 사용되는 방법은 3가지가 있다
1) Gene fragment recruitment
- Metagenome read 혹은 contig를 database에 mapping하는 것이다.
- Full-length gene sequence와 동일하거나 유사한 metagenomic sequences는 유전자의 representative subsequences로 간주된다.
- Read가 database에 있는 sequence와 비교하는 알고리즘을 사용하기 때문에 속도가 빠른 high-throughput gene prediction 방법이다.
- 새로운 유전자이거나 database에서 underrepresentative할 경우 적합하지 않다.
2) Protein family classification
- Metagenomic read를 가능한 모든 protein coding frames로 번역하고 생성된 peptide 각각을 sequence alignment를 통해 protein sequence database와 비교한다.
- Alignment를 시킨 후 database의 단백질과 상동성을 나타내는 번역된 peptide를 encoding하는 metagenomic sequence를 식별하는 분석이 진행된다.
- USEARCH, RAPsearch 또는 lastp와 같은 blastp 또는 fast blast 알고리즘을 사용하여 단백질 sequence alignment를 진행할 수 있다.
3) de novo gene prediction
- 새로운 유전자를 식별이 가능하다.
- 미생물 유전자의 길이, codon의 사용, GC bias 등의 다양한 특성을 사용해 훈련된 유전자 예측 모델을 사용하여 metagenomic read 또는 contig가 유전자를 포함하고 있는지, reference database의 sequence와 유사성에 의존하지는 않는지 등을 평가한다.
- 이 방법을 통해 다른 미생물 유전자와 공통된 특성을 공유하지만 기존의 알려진 유전자와는 크게 다를 수 있는 유전자를 식별할 수 있었다.
- 대표적인 tools로는 MetaGene, Glimmer-MG, MetaGeneMark, FragGeneScan, Orphelia, MetaGun 등이 있다.
7 - 2) Functional annotation by protein family classification
- Protein family란 진화적으로 관련된 protein sequences, 또는 protein domain families의 subsequences이다.
- 예측된 metagenomic coding sequences에 functional annotation을 하는 가장 일반적인 방법으로 genome sequencing projects를 통해 알려진 full-length protein sequences와 비교를 함으로써 특정된다.
- Protein family끼리는 서로 공통 조상을 공유하기 때문에 유사한 biological function을 암호화할 것이라는 가설을 바탕으로 진행된다. Metagenomic sequence가 해당 family와 homolog하다고 결정될 경우 (해당 family의 member로 분류될 경우), 해당 sequence가 family의 function을 암호화한다고 추론한다.
- Metagenomic sequence가 모든 proteins 혹은 models와 비교되면 3가지 결과 중 하나가 나타난다.
- A single family (가장 hit가 높은 family)
- A series of families (예로, 모든 families가 유의미한 classification score를 보임)
- No family (새로운 단백질이거나 매우 높은 다양성을 가지거나 가짜, 3가지 중 하나)
- A single family (가장 hit가 높은 family)
- Functional annotation에 사용되는 DB는 크게 2가지로 나눌 수 있다
- Sequence database
- 상대적으로 빠르며 database의 sequence와 밀접하게 관련된 read에 대해 구체적인 hit를 측정할 수 있다.
- 대표적인 예로 KEGG, MetaCyc, EggNOG 등이 있다.
- 상대적으로 빠르며 database의 sequence와 밀접하게 관련된 read에 대해 구체적인 hit를 측정할 수 있다.
- HMM database
- 매우 짧은 sequence에 대해서는 정밀도가 낮을 수 있다.
- 대표적인 예로 Phylofacts, SiftingFamilies 등이 있다.
- 매우 짧은 sequence에 대해서는 정밀도가 낮을 수 있다.
- Sequence database
- Metagenomic read의 protein family classification을 통한 function prediction의 주의점
- Metagenome에서 암호화된 functional diversity는 community의 functional activity와 유사할 뿐이다.
유전자가 있다고 해서 sampling 시점에서 발현되는 것이 아니기 때문이다. Comparative metagenomic과 metatranscriptomic analyses는 transcriptional level에서 community 간의 차이가 종종 genomic level에서 반영된다는 것을 나타내며, 이는 metagenome이 activity에 대한 의미있는 대체제라는 것을 시사한다. - 대부분의 database에는 알려진 functional annotation이 없는 families가 포함되어 있다.
이러한 families의 homologs로 결정된 metagenomic reads는 function으로 간주되지 않는다. - sequence에 annotation을 하기 위해 사용된 protein family database는 phylogenetic biases에 영향을 받을 수 있다.
특정 community는 다른 community보다 불균형적으로 더 정확하거나 더 엄격하게 annotation될 수 있다. 또한 각 database가 family를 식별하고 functional annotation을 하기 위해 다른 알고리즘을 사용하기 때문에 서로 다른 functional profile을 생성할 수 있다. - 이 방법은 function이 진화적으로 정적이라는 가정으로 진행된다.
진화적으로 가소성을 가진 function은 function을 예층하는 특수성을 약화시키기 때문이다. - 자연계에는 현재 database에 기술된 것보다 더 많은 protein과 function이 있을 수 있다.
- Metagenome에서 암호화된 functional diversity는 community의 functional activity와 유사할 뿐이다.

Reference
- An introduction to the analysis of shotgun metagenomic data, Frontiers, Thomas J. Sharpton
- Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics, Computational and Structural Biotechnology Journal, KarelSedlar, Kristyna Kupkova, Ivo, Provaznik